Why The New FDA Update Matters Right Now for MDMs
The FDA’s Final Cybersecurity Guidance of June 2025 introduces significant new requirements under the FD&C Act, making cybersecurity a lifecycle obligation. Section 7 provides the most detailed interpretation to date.
The update consolidates all previous cybersecurity guidance documents and specifies how medical device manufacturers (MDMs) must address both premarket and postmarket risks, including the systems surrounding the device.
For your convenience, we summarized the new requirements of the cybersecurity mandate for MDMs to move forward proactively.

Going Back to the 524B Cybersecurity Mandate
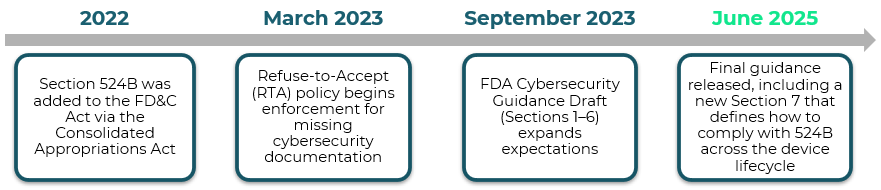
Section 524B of the FD&C Act requires MDMs to include cybersecurity information in their premarket approval applications to demonstrate that their devices meet security requirements.
A study showed that 53% of connected medical and healthcare IoT devices have at least one unpatched critical vulnerability.
MDMs must show that their products are designed with security top of mind, from initial development through deployment, to protect patient safety. Section 524B has become the new roadmap for cybersecurity compliance in medical devices.
The table below illustrates the full timeline of Section 524B:
What’s New in the FDA’s Guidelines: Key Highlights
What Qualifies as a Cyber Device?
Section 7 further expands the scope to include:
- Internet-connected devices, such as Infusion pumps with remote monitoring, cardiac implantable electronic devices (CIEDs) with cloud synchronization, hospital beds that transmit usage data over Wi-Fi and more.
- Devices with USB ports or wireless modules such as Insulin pumps with Bluetooth connectivity, diagnostic imaging systems with USB data export, wearable ECG monitors with mobile app pairing and more.
- Systems with programmable logic components such as Robotic surgical systems, radiation therapy machines with configurable treatment plans, smart ventilators with field-updatable firmware and more.
Expanded View: Securing the Healthcare Ecosystem
Compliance now includes related systems, such as:
- Software/firmware update servers.
- Hospital networks or integration points.
- Stringent device design controls.
- Manufacturer-controlled assets of the medical device system reaching end of life (EOL) or support.
Manufacturers must “establish and maintain procedures for validating the device design.” MDMs should design devices to limit the potential impact of vulnerabilities by enforcing a secure default configuration and notifying users when anomalous device behavior is detected.
There are other security barriers for MDMs, such as changes to authentication or encryption algorithms, which may delay product development or trigger additional regulatory review late in the product lifecycle, ultimately impacting device time-to-market and key stakeholders. A thorough risk assessment should document any risk mitigations to prevent device hazards in a premarket submission.
Security Updates to Existing Devices
- Major changes: Cryptography and interfaces.
The new regulation suggests that MDMs follow NIST guidelines for cryptography, such as FIPS 140-3, to ensure security throughout the device’s lifecycle. It also recommends avoiding device serial numbers as keys in the event of a recall disclosure, where exposure may compromise the confidentiality and integrity of the device’s cryptographic systems.
Manufacturers must disclose all communication interfaces of medical devices, especially those involving third-party software, as they may expose sensitive patient data and allow unauthorized access. The FDA also recommends that MDMs include detailed diagrams illustrating system architecture, interfaces, and communication protocols for full traceability. - Minor or non-security changes: Streamlined paths.
MDMs should consider the end-to-end path of device design to minimize additional risk and protect authentication and authorization procedures. The design diagrams should map data flows, identify trust boundaries, and highlight components handling credentials or tokens containing sensitive data.
The section also requires MDMs to include a description of any current critical vulnerabilities that could pose uncontrolled risks.
Controlling user access is essential in preventing cyber risks. The section highlighted security control categories, such as authentication and authorization, among other areas.
Authentication best practices:
- Implement MFA to permit privileged device access to system administrators.
- Avoid using hardcoded passwords.
- Use telemetry to verify the authenticity of information originating from the device.
- Limit session duration for external communications.
Authorization best practices:
- Limit authorized access to devices,
- Implement the principle of least privilege, and assign permissions by roles only,
- Design devices to “deny by default”,
- Employ automatic session termination based on predefined time limits within the medical device system.
Premarket Planning Requirements for MDMs
Section 7 mandates that premarket submissions must include:
- A Coordinated Vulnerability Disclosure (CVD) policy.
- Code review methods, including SAST and DAST to parse external data.
- Defined patch/update timelines, aligned to risk level.
- Event detection and logging.
- A threat model and risk management plan that is maintained over time, continuously. updated based on new threats and used during postmarket surveillance.
- Supply chain cybersecurity risk management.
- An SBOM (Software Bill Of Material) of all commercial, open-source, and off-the-shelf software components.
Section 524B requires manufacturers of cyber devices to provide an SBOM, which enables manufacturers to track all software components, dependencies, and potential vulnerabilities within the device, ensuring full transparency throughout the product lifecycle. Every component listed in the SBOM should include metadata such as version, supplier, and license type for accurate tracking, vulnerability assessment, and compliance.
Risk-Based Update Requirements
The FDA now classifies security risks as either:
- Uncontrolled: Requires immediate updates. The FDA defines uncontrolled risk as “unacceptable residual risk of patient harm due to inadequate compensating controls and risk mitigations.” It also recommends that manufacturers implement a cybersecurity vulnerability assessment tool or scoring system, such as the CVSS, to categorize, prioritize, and mitigate threats.
The FDA suggests that manufacturers disclose vulnerabilities within 30 days of discovery and communicate relevant information to their customers. - Controlled: Follows a scheduled maintenance window. The FDA’s best practices recommend implementing a Secure Product Development Framework (SPDF) to minimize risk throughout the product lifecycle. Integrating SPDF processes early in the design phase helps MDMs avoid re-engineering devices later, particularly when connectivity features are added after market distribution.
Exceeding the FDA’s Latest Regulatory Updates with C2A Security
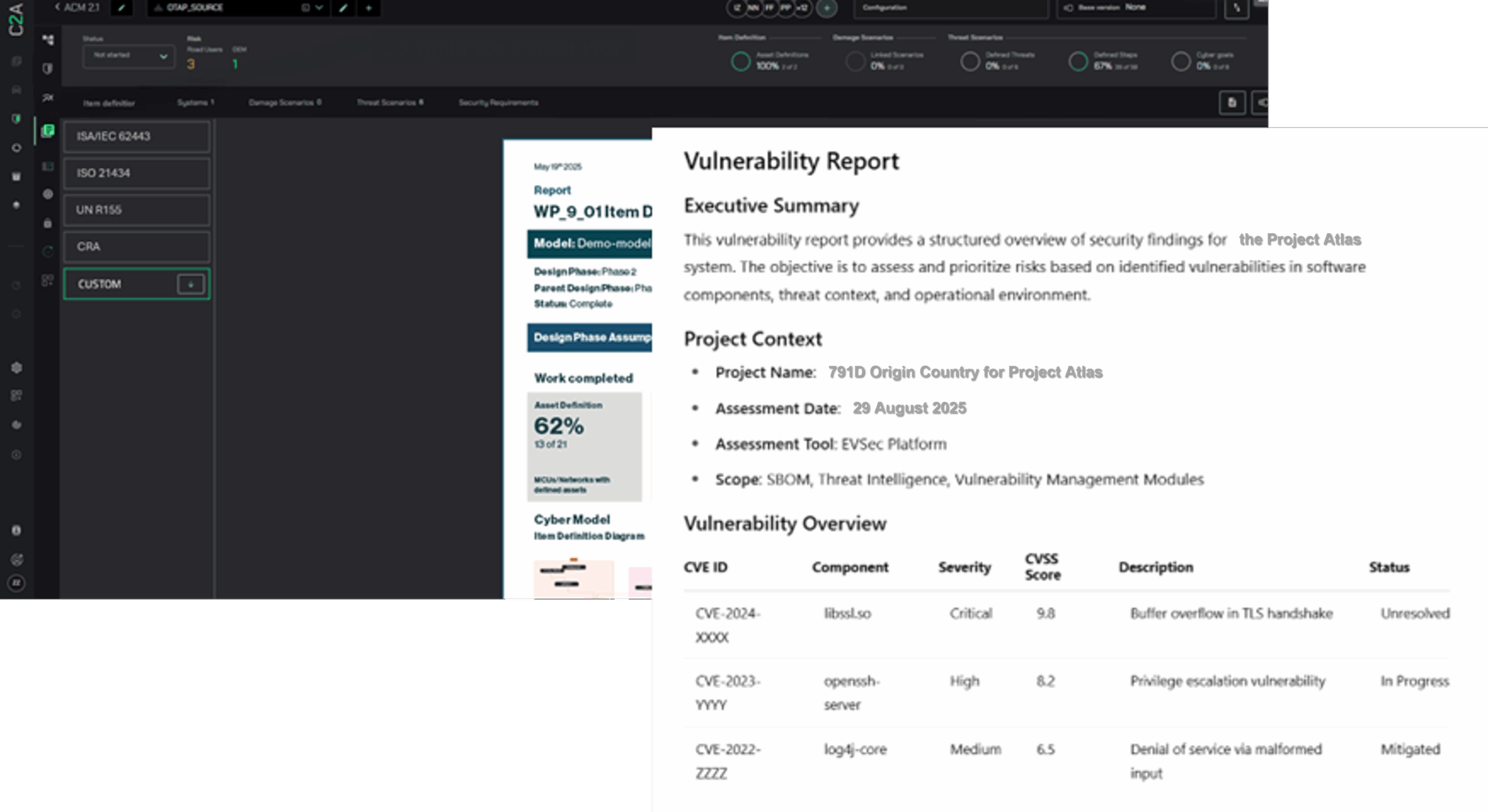
C2A Security’s EVSec platform supports medical device companies in meeting the FDA’s latest cybersecurity requirements by:
- Maintaining a live threat and risk model throughout the Total Product Lifecycle (TPLC). Ongoing TARA.
- Automating SBOM management, CVD workflows, and update tracking.
- Mapping vulnerabilities and threats to system architecture, regulatory. mandates, and mitigation strategies.
- Providing compliance-ready evidence for premarket submissions and postmarket audits.
- Enabling teams to triage, collaborate, and track incident responses across all stakeholders.
Schedule a demo to learn how C2A Security can help you stay ahead of the FDA’s latest regulatory requirements for medical devices.